第 5 章 化学平衡导论
摘要:本章介绍了混合物和理想溶液的平衡;介绍了平衡常数、它与吉布斯自由能的关系以及它对温度的依赖性;区分了均相平衡和非均相平衡;并介绍了固-气、液-气和矿物-溶液平衡,包括亨利定律和溶度积。
关键词:平衡常数 · 反应商 · 溶度积 · 范特霍夫方程
我们先前的热力学课程为我们提供了判断在地球表面条件下,一个反应是否自发进行的工具:判断反应的吉布斯自由能变化是负值还是正值。例如,氯化钠(盐)的溶解是一个吸热过程(吸收热量并使水温下降;\(\Delta H^0 > 0\)),但它自发发生,因为它的吉布斯自由能变(\(\Delta G^0\))为负值,约为\(-9 \text{kJ} \cdot \text{mol}^{-1}\)。反应的自发性并不意味着反应一定会完全进行,也就是说,平衡状态下并非所有反应物都会转化为生成物。当氯化钠加入水中时,确实会发生溶解,但在1千克水约加入357克氯化钠后,溶解过程停止,不再观察到进一步溶解。如果加入超过357克氯化钠,它将开始沉淀(逆反应)。这时水-氯化钠系统达到了平衡状态(equilibrium),即反应物和生成物的浓度在一段时间内保持不变。平衡状态的形成是因为正向反应(从反应物到生成物)和逆向反应(从生成物到反应物)的速率相等,且浓度不再发生变化。在地球系统中,许多反应可以通过平衡方法描述或进行近似描述:水与空气之间的气体交换、质子转移(酸碱化学)、电子转移(氧化还原反应)以及矿物的沉淀与溶解。例如,碳酸钙的溶解导致喀斯特地貌的形成,而碳酸钙的沉淀则导致溶剂中钾乳石和石笋的形成。
因此,深入理解并进一步学习预测平衡的定量工具是很有用的。本书中,我们重点讨论理想溶液和地球表面条件下的平衡,非理想溶液以及在岩浆岩和变质岩形成过程中,高温高压条件下的平衡将有待后续的专业课程讲解。在介绍平衡常数及其与吉布斯自由能的关系后,我们将重点讨论涉及矿物、酸碱、氧化还原中的平衡。它们都在理想溶液的范畴中讨论。
5.1 平衡常数及其与吉布斯自由能的关系
二氧化碳(\(\mathrm{NO_2}\))是一种大气中的气体,由引擎燃烧(比如汽车、公交车、卡车)产生。它是一种深棕色气体,参与形成了城市上空的雾霾。在大气中,它与四氧化二氮(无色气体)处于平衡状态:
\(\mathrm{NO_2(g)}\)转化为\(\mathrm{N_2O_4(g)}\)会导致颜色逐渐褪去,直到建立平衡状态。这一反应在实验室中得到了充分研究。不论初始条件如何(仅有\(\mathrm{NO_2(g)}\)或仅有\(\mathrm{N_2O_4(g)}\)或高浓度\(\mathrm{NO_2(g)}\)与低浓度\(\mathrm{N_2O_4(g)}\),或反之),当以生成物与反应物浓度比表示反应混合物的最终组成时,结果总是相同的。这就是质量作用定律(the law of mass action):具体而言,在25°C和1 bar下,计算出的浓度比\(\frac{\mathrm{N_2O_4(g)}}{\mathrm{NO_2(g)}^2}\)约为216,无论初始条件如何。具体来说,对于\(\alpha\) mol物质A和\(\beta\) mol物质B反应生成\(\gamma\) mol物质C和\(\delta\) mol物质D的反应:
有反应商(reaction quotient)\(Q\):
反应商是无量纲的注①,也是普适性的,即无论系统是否处于平衡状态,都可以计算。当系统处于平衡时,\(Q\)的值称为平衡常数(equilibrium constant)\(K\)注②:
\(K\)的值表达了系统的组成,即平衡时反应物和生成物的浓度或分压(考虑到反应的化学计量数),并且在一定的温度和压力下具有恒定值。平衡常数没有单位,因为各项单位会相互抵消。逆向反应会导致\(K\)值的倒置,而将反应的系数(\(\alpha, \beta, \gamma, \delta\))乘以一个公共因子,则平衡常数会提高到相应的因子。如果\(K\)值非常大,平衡状态下的混合物将主要由生成物C和D组成。如果\(K \ll 1\),则反应物将在混合物中占主导地位,而当\(K\)值接近1时,反应物和生成物的量大致相等,当然,这也取决于反应的化学计量数。
K>>1 生成物占主导地位;
K≈1 反应物和生成物的量大致相等;
K << 1 反应物占主导地位。
比较反应商\(Q\)和平衡常数\(K\)可以提供关于变化方向的信息,即判断一个过程是否自发。如果\(Q > K\),则净反应将从右向左进行(即从生成物转化为反应物,逆反应是自发的);如果\(Q < K\),则反应将从左向右进行(即反应物转化为生成物,正反应是自发的)。如果\(Q = K\),则反应处于平衡状态。
Q > K 逆反应自发;
Q < K 正反应自发;
Q = K 反应处于平衡状态。
在前面的热力学课程中,我们已经看到,自发性与恒压恒温下的吉布斯自由能变化有关;因此,反应商\(Q\)、平衡常数\(K\)和吉布斯自由能变化(\(\Delta_r G\))之间应该存在一种关系。回到式5.1,对于气相反应\(\mathrm{NO_2(g)}\)转化为\(\mathrm{N_2O_4(g)}\)的吉布斯自由能变化(\(\Delta_r G\)):
从先前的热力学课程中(式4.12),我们知道:
其中,\(R\)是普适气体常数,\(T\)是绝对温度,\(\Delta G^\circ\)是纯气体的标准吉布斯自由能变,\(p_g\)是气体g的分压,\(p_0\)是总压力,取1 bar。结合式5.5和式5.6,我们可以得到:
用\(\Delta_r G^\circ = \Delta G^\circ (\mathrm{N_2O_4(g)}) - 2 \times \Delta G^\circ (\mathrm{NO_2(g)})\),我们可以整理式5.7如下:
因此,反应的吉布斯自由能变\(\Delta_r G\)包含了与反应物和生成物的分压无关的标准吉布斯自由能变项\(\Delta_r G^\circ\),以及一个项\(RT\ln \left[\left(\frac{p_{\mathrm{N_2O_4}}}{p_0}\right)/\left(\frac{p_{\mathrm{NO_2}}}{p_0}\right)^2\right]\),该项依赖于反应物和生成物的分压。
此外,如果用反应物和生成物的分压以及总压力\(p_0 = 1 \text{bar}\)来表示,\(RT\ln \left[\left(\frac{p_{\mathrm{N_2O_4}}}{p_0}\right)/\left(\frac{p_{\mathrm{NO_2}}}{p_0}\right)^2\right]\)就是式5.1的反应商\(Q\)。
将这一点推广到任何反应,我们可以得到:
即反应的吉布斯自由能变是反应的标准吉布斯自由能变\(\Delta_r G^\circ\)与项\(RT\ln Q\)之和,其中后者为实际混合物的组成提供了修正项。
作为一个例子,考虑式5.1和一个在298 K下的反应容器,其中\(\mathrm{NO_2(g)}\)的分压为0.350 bar,\(\mathrm{N_2O_4(g)}\)的分压为0.65 bar。两种气体的标准生成吉布斯自由能\(\Delta G^\circ\)分别为51.3 kJ·mol\(^{-1}\)和99.8 kJ·mol\(^{-1}\)。
\(\Delta_r G \neq 0\),因此,系统不处于平衡状态。具体来说,净反应向反应物方向进行,即向\(\mathrm{NO_2(g)}\)方向进行,因为\(\Delta_r G > 0\)。使用反应商\(Q\)和平衡常数\(K\)也可以得出相同的结论。
在平衡状态下,反应商等于平衡常数\((Q = K)\),此时反应的吉布斯自由能变应为零\((\Delta_r G = 0)\)。因此,
故
或注③
式5.10a和式5.10b是普适性的,适用于所有类型的反应。一方面,它们提供了热力学数据和理论之间的直接联系,另一方面它们与平衡时混合物的组成相关。然而,式5.10a和式5.10b意味着混合物的组成是以活度而非浓度表示的。在本课程中,我们的讨论将仅限于理想溶液,即忽略离子对和离子-溶剂之间的相互作用,且我们将平衡常数和反应商用浓度\([\mathrm{c}]\)来表示,而不是活度\(a\),其中气体的浓度\([\mathrm{c}] = p_g / p_0 (p_0 = 1 \text{bar})\),溶质的浓度\([\mathrm{c}] = c_a / c_0 (c_0 = 1 \text{mol} \cdot \text{L}^{-1})\),固体或液体介质的浓度\([\mathrm{c}] = 1\)。 活度和浓度通过活度系数相关联。然而,使用浓度而非活度意味着平衡常数需要单位。
平衡态混合物的组成是依赖于温度的。有两种方法可以修正平衡常数以适应较低或较高的温度。第一种方法是使用式5.10a和式5.10b,将平衡常数\(K\)与在特定温度下计算的反应吉布斯自由能变化\(\Delta_r G^\circ\)关联起来,该计算基于反应的熵\((\Delta_r S^\circ)\)和焓\((\Delta_r H^\circ)\)。
另一种方法是使用范特霍夫方程(van't Hoff equation)。联立式5.10a:\(\Delta_r G^\circ = -RT \ln K\)和式4.3:\(\Delta_r G^\circ = \Delta_r H^\circ - T \Delta_r S^\circ\),整理后我们就得到了它:
或者是它的另外一种形式,应用于温度由\(T_1\)到\(T_2\)的变化中:
其中,\(T_1\)通常为298 K(SATP)。如果反应是放热反应\((\Delta_r H^\circ < 0)\),则随着温度的升高,\(K\)值会减小,平衡会向反应物方向移动,即向左移动。相反,对于吸热反应\((\Delta_r H^\circ > 0)\),平衡会向右移动,这样,\(K\)值随着温度的升高而增大。
5.2 异相平衡与矿物溶解平衡
平衡可以在所有反应物和生成物处于同一相时建立,即同相平衡(homogenous equilibrium),也可以在涉及多个相的情况下建立,即异相平衡(heterogenous equilibrium)。溶液中的酸碱反应是同相平衡的一个例子,而涉及固体和液相的矿物溶解沉淀反应则是异相平衡的例子。本节将介绍三种类型的异相平衡(固-气、液-气和矿物-溶液)。同相酸碱平衡将在下一章中讨论。
固-气平衡
考虑如下从石灰岩(\(\mathrm{CaCO_3(s)}\))生产石灰(\(\mathrm{CaO(s)}\))的反应:
这是一个异相反应,其平衡常数 \(K\) 是
注意,\([...]\)用来表示浓度而非活度。固相和液相可以从异相平衡的质量作用定律中排除,因为它们的(活度)值为1。因此,在包含固相 \(\mathrm{CaO}\) 和 \(\mathrm{CaCO_3}\) 的系统中,二氧化碳气体的平衡浓度与这些固相的数量无关,只要它们都存在。
液-气平衡
水中的溶解度是另一个异相平衡的例子。气体在水中的溶解度使用亨利定律注④(Henry's law)来描述:
其中,\(c_{\mathrm{gas}}\) 是气体在水中的平衡浓度(\(\mathrm{mol\,kg^{-1}}\)),\(p_{\mathrm{gas}}\) 是气体的分压(\(\mathrm{atm}\)),\(K_H\) 是亨利定律常数(\(\mathrm{mol\,kg^{-1}\,atm^{-1}}\))。亨利定律常数是水的温度和盐度的函数注⑤。
考虑氧气在水中的溶解:
对应的平衡常数是
图 5.1 显示了溶解氧浓度随温度变化的曲线,分别对应淡水和海水的情况。氧气及大多数其他气体在淡水中的溶解度大于海水,这是由于盐析效应(salting out effect)。随着水温升高,水中溶解氧的浓度会降低。因此,处于与大气平衡状态的冷水比暖水具有更高的溶氧量。
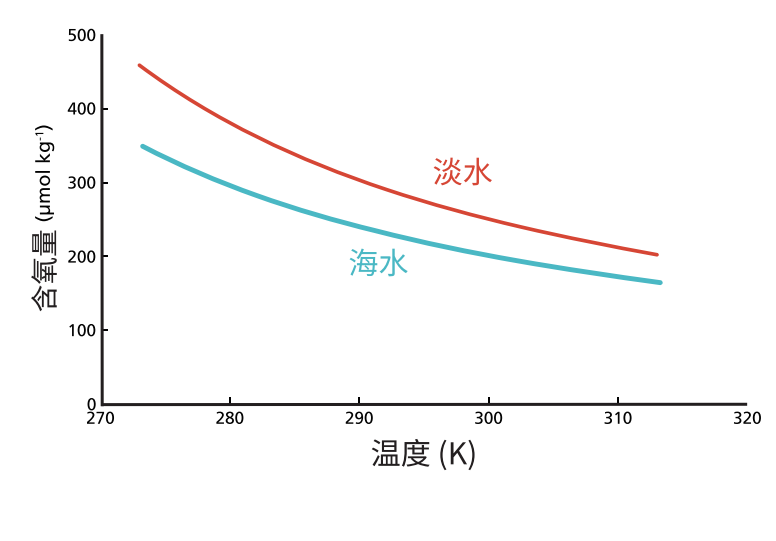
图5.1 氧气在淡水与咸水中的溶解度是温度的函数
矿物-溶液平衡
某些矿物的溶解度限制了天然水的组成成分,反之,溶液的组成成分可以决定矿物是溶解还是沉淀。矿物平衡通常以溶解反应的形式表示(即矿物在反应方程的左侧,离子在右侧),并通过溶解度积(solubility product)来表征。比如,萤石(\(\mathrm{CaF_2(s)}\))的溶解度积基于以下反应:
其平衡常数是
其中,脚标 \(\mathrm{eq}\) 表示这些是平衡时的浓度。回想一下,固相的摩尔分数为1,我们可以定义萤石的溶解度积为:
溶解度积 \(K_{sp}\) 是固相(矿物)溶解这一特殊情况下的平衡常数 \(K\)。这一概念可以推广为:
而
矿物的沉淀和溶解取决于该矿物的饱和状态(saturation state)。对于以方解石形式存在的碳酸钙溶解:
我们首先定义离子积(Ion Product, IP),它是矿物平衡中反应商 \(Q\) 的一种特殊形式:
这样,它的单位是 \(\mathrm{mol^{2}\,kg^{-2}}\),那么它的饱和度(degree of saturation)或饱和状态 (\(\Omega\)) 是:
如果 \(\Omega < 1\),即在饱和度低的水中,方解石将溶解;如果 \(\Omega > 1\),即水体相对于该矿物过饱和,则方解石会从溶液中沉淀出来。
注释
1 “无量纲的”(dimensionless)是指没有任何单位的量。它通常表示一个纯粹的数字比例或比值, 能够表达两者之间的关系,但不依赖于具体的物理单位。
2 平衡常数既可以用浓度表示,也可以用平衡分压表示。
3 exp 指的是以自然对数 e 为底的指数式,也就是 exp (x)= ex。
4 亨利(1775—1836),英国化学家、医生。
5 某些气体的常数:
氧气(O₂):\(769.2\ \mathrm{mol\ kg^{-1}\ atm^{-1}}\);
二氧化碳(CO₂):\(29.4\ \mathrm{mol\ kg^{-1}\ atm^{-1}}\);
氢气(H₂):\(1282.1\ \mathrm{mol\ kg^{-1}\ atm^{-1}}\)。